Summary
1) Managed the code for data processing, modeling, and hypothesis testing.
2) Helped write the methodology/statistics part of the paper (Rodriguez-Duboc et al., 2025).
3) Open-sourced the resulting code on GitHub and registered it on Zenodo.
Data
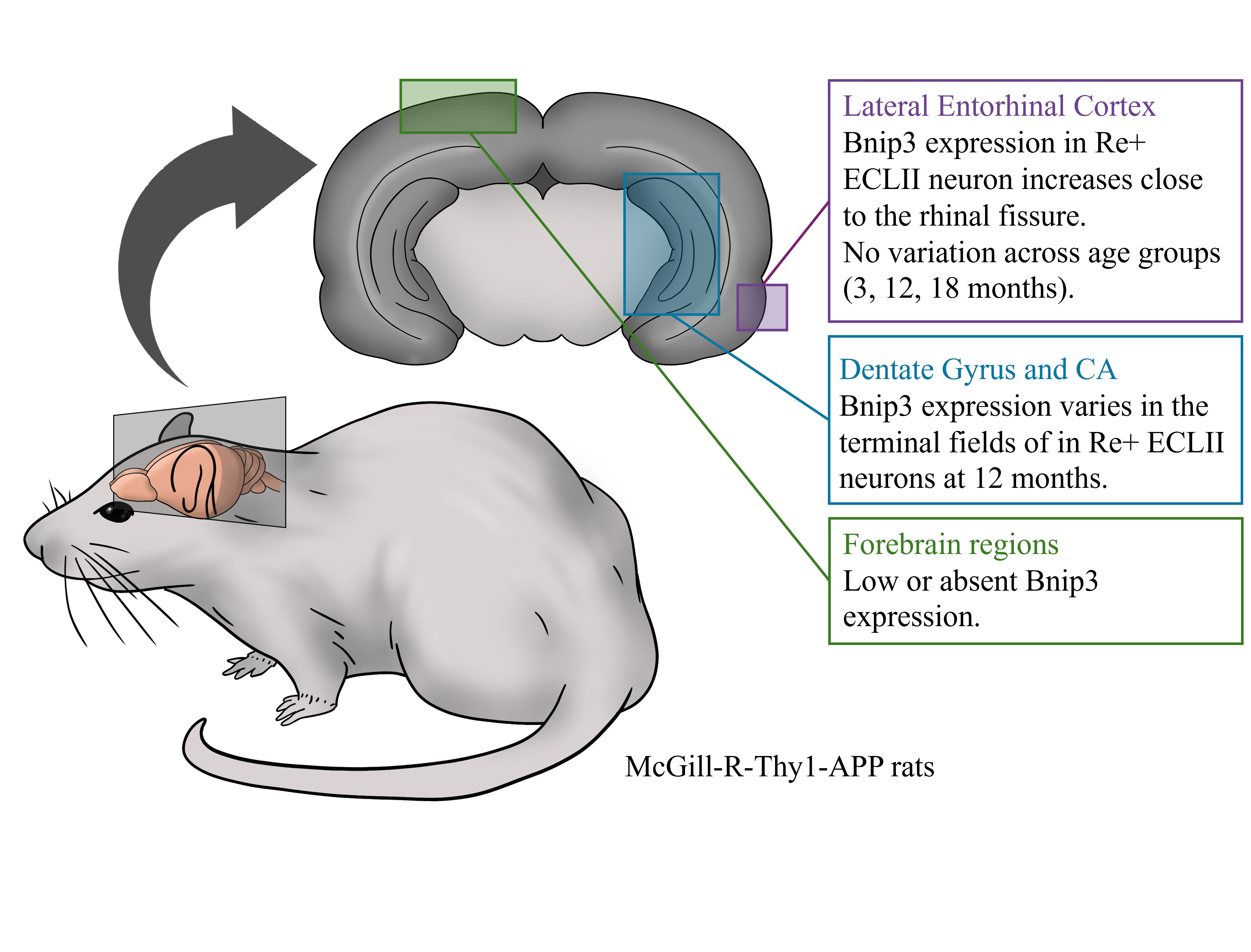
The project analyzed brightfield immunohistochemistry (IHC) data quantified as optical density (OD) measurements from brain tissue sections. Data was collected from 24 rats (12 AD model, 12 controls) across three age groups (3, 12, 18 months) to measure BNIP3 protein expression in neuronal soma and axonal projections. Each animal contributed 7-8 tissue sections covering specific brain regions (entorhinal cortex, CA3, dentate gyrus), with background correction applied by subtracting deep layer measurements from raw OD values. The dataset included hierarchical structure with measurements nested within sections, sections within animals, creating pseudo-replication that required mixed-effects modeling approaches.
Data preprocessing involved standardizing variable names and metadata using a data dictionary to ensure consistent analysis workflows. The final dataset captured both cellular (soma) and projection field (molecular layer) measurements across genotype, age, and brain region factors.
Models
Optical Density (OD) data was modeled using a Gamma distribution to account for the data’s strict positivity and positive skewness, with a log link function.
Generalized Linear Mixed Models (GLMMs) were used to model the data, using random effects to account for pseudo-replication and the hierarchical nature of the data. For all models, categorical predictors were dummy-coded with a reference level: genotype (Wild-Type) and age (3 months). Models were fitted using the glmmTMB package (Brooks et al., 2017).
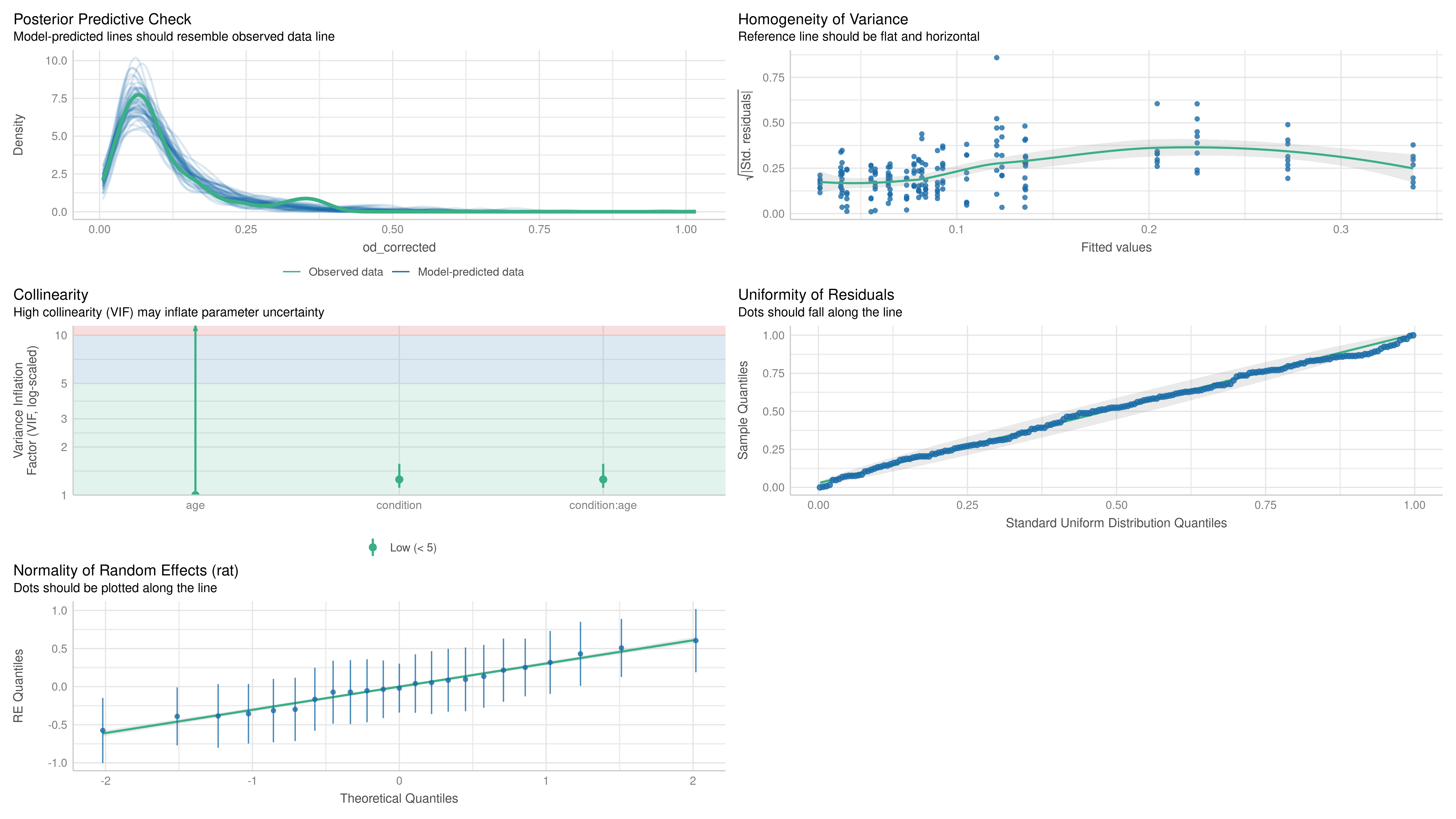
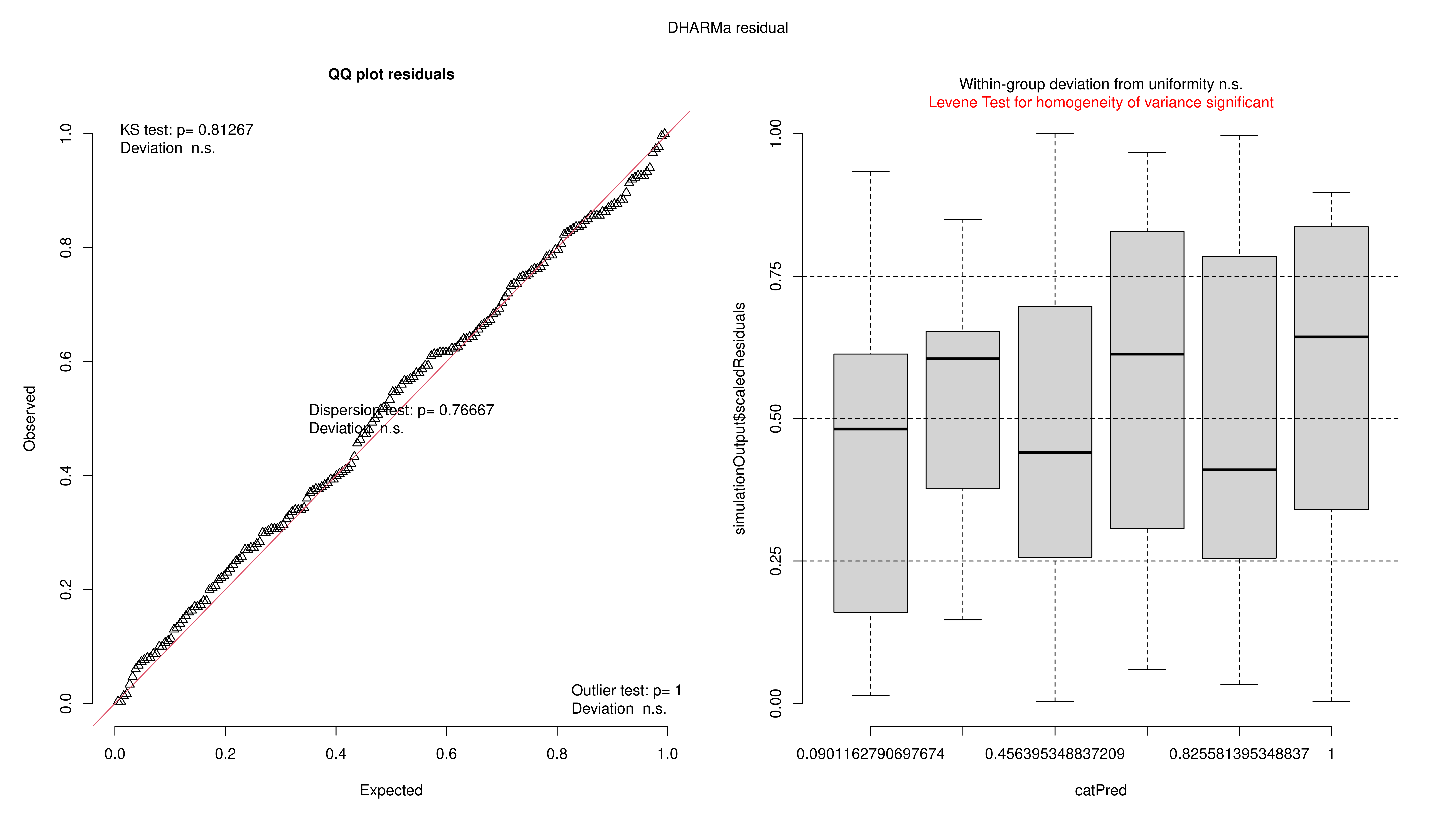
Model fit was assessed using a combination of plots (normality, homoscedasticity, normality of residuals, collinearity), statistical tests (Levenne, KS, dispersion), and posterior predictive checks on simple statistics (min, max, mean, sd) using simulated residuals. Model fit was assessed using the DHARMa (Hartig, 2022) and performance (Lüdecke et al., 2021) packages.
Two models were fitted on the OD data, one for the soma and one for the molecular layer (terminal fields):
We modeled the OD of individual neuronal somata () using genotype, age, and brain area (LEC vs. MEC) as fixed effects, along with all their interactions. Random intercepts were included for each rat (j) and for each tissue section (k) nested within each rat.
Model equation
Model fit
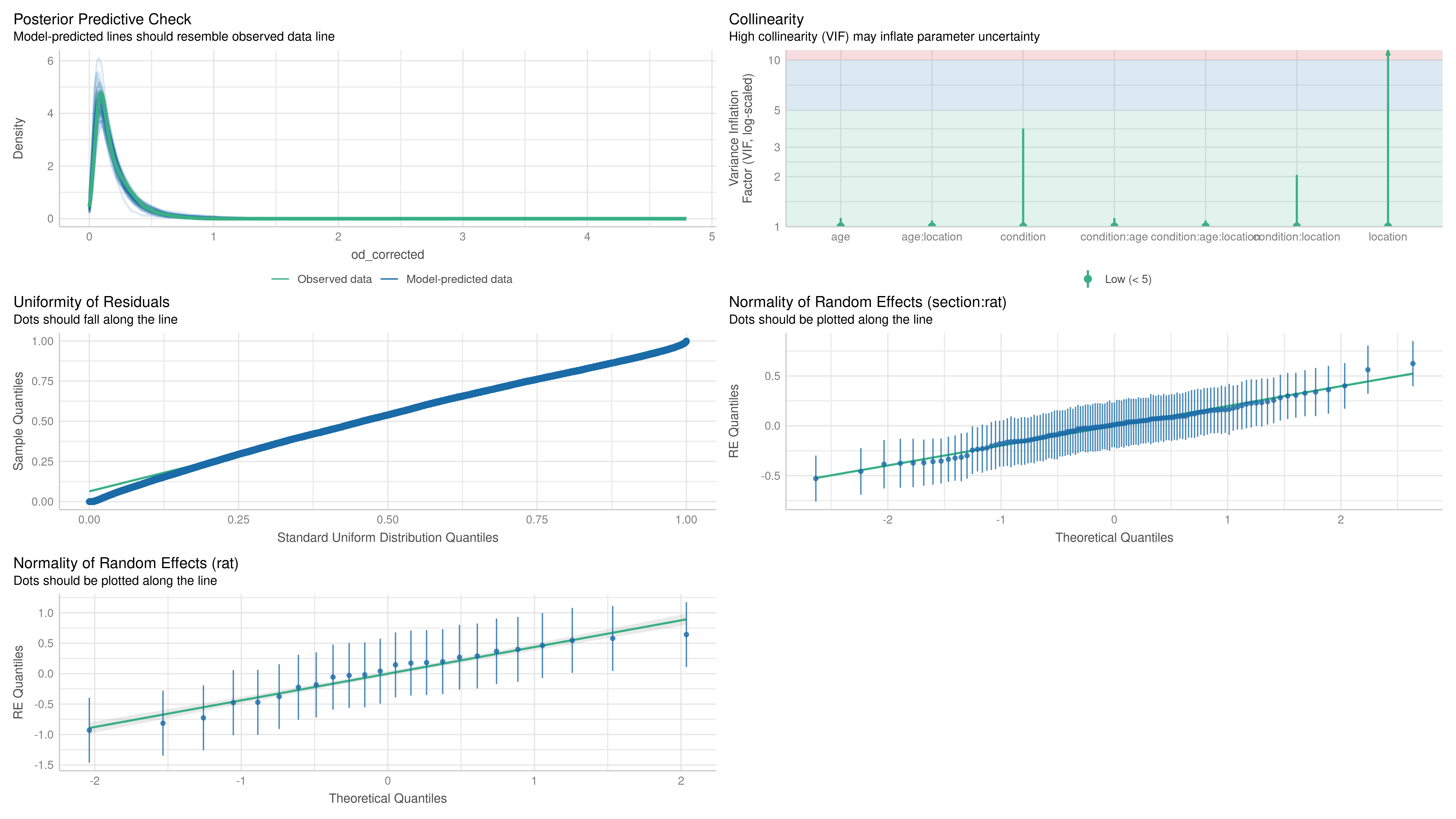
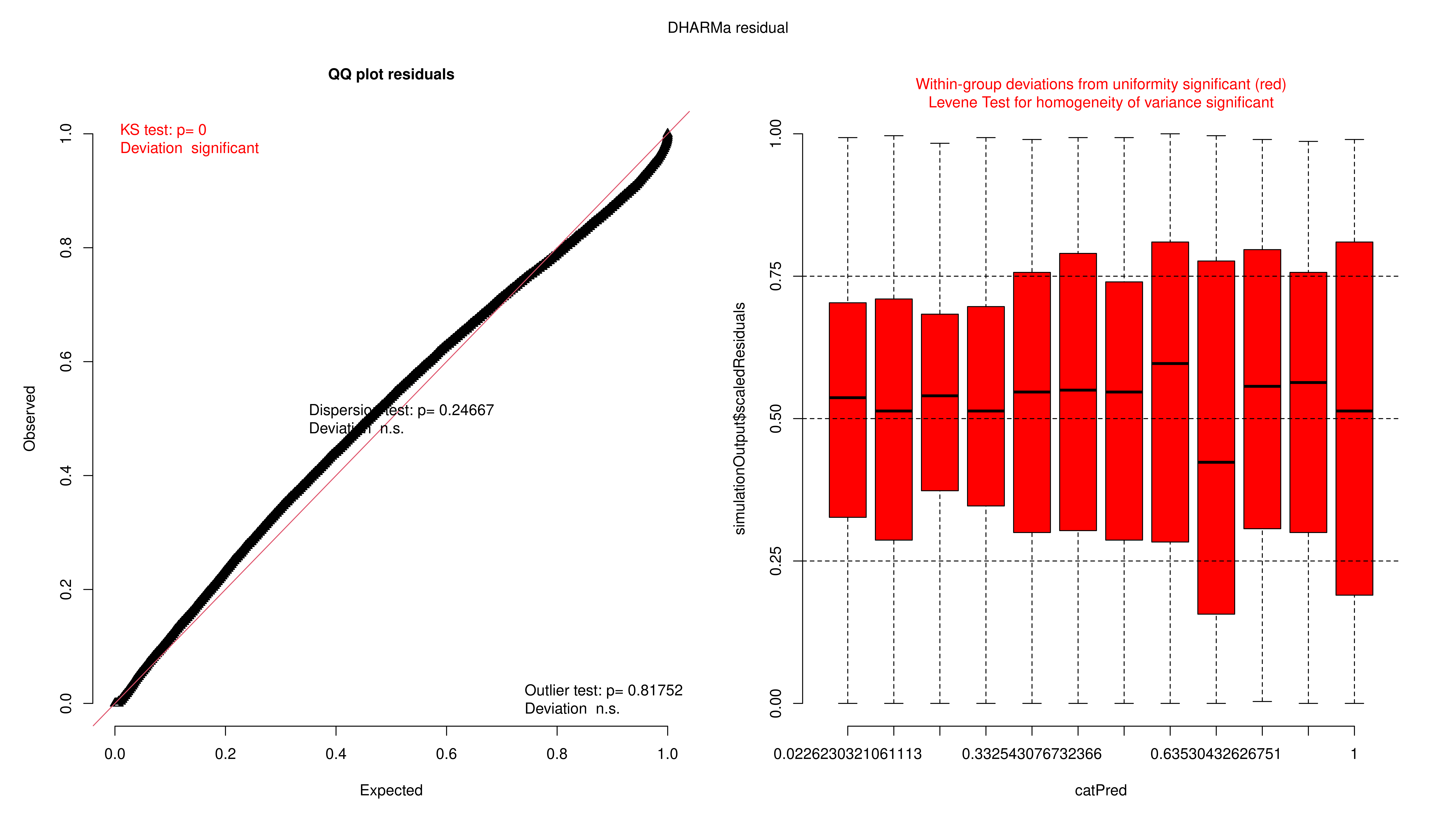
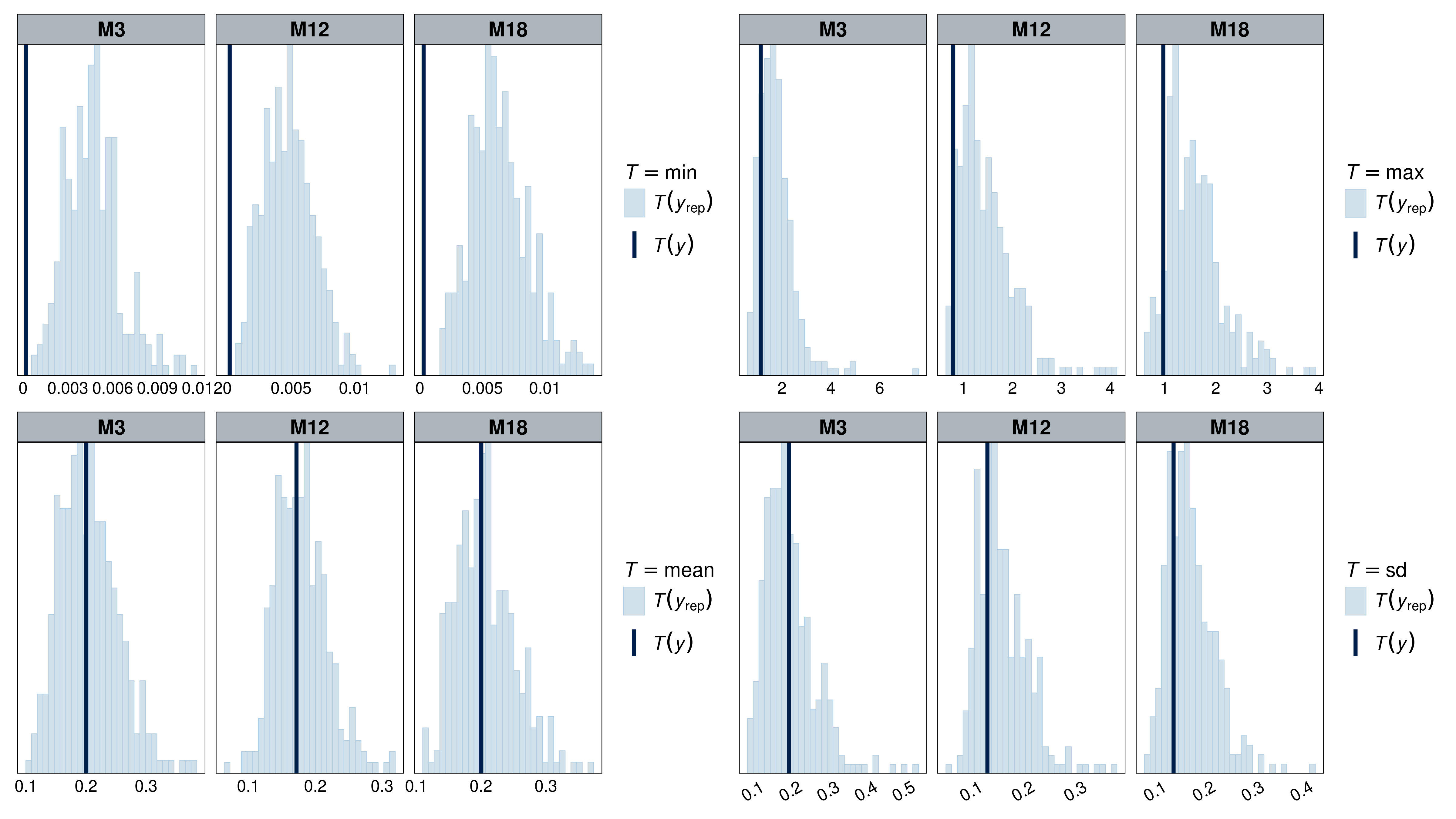
To model the OD of the terminal fields (), the fixed effects included genotype, age, and their interaction. A random intercept for each rat (j) was included to account for inter-animal variability.
Model equation
glmmTMB(
od_corrected ~ genotype * age + (1 | rat),
family = Gamma("log"),
data = od_data$ml,
REML = TRUE
)Model fit
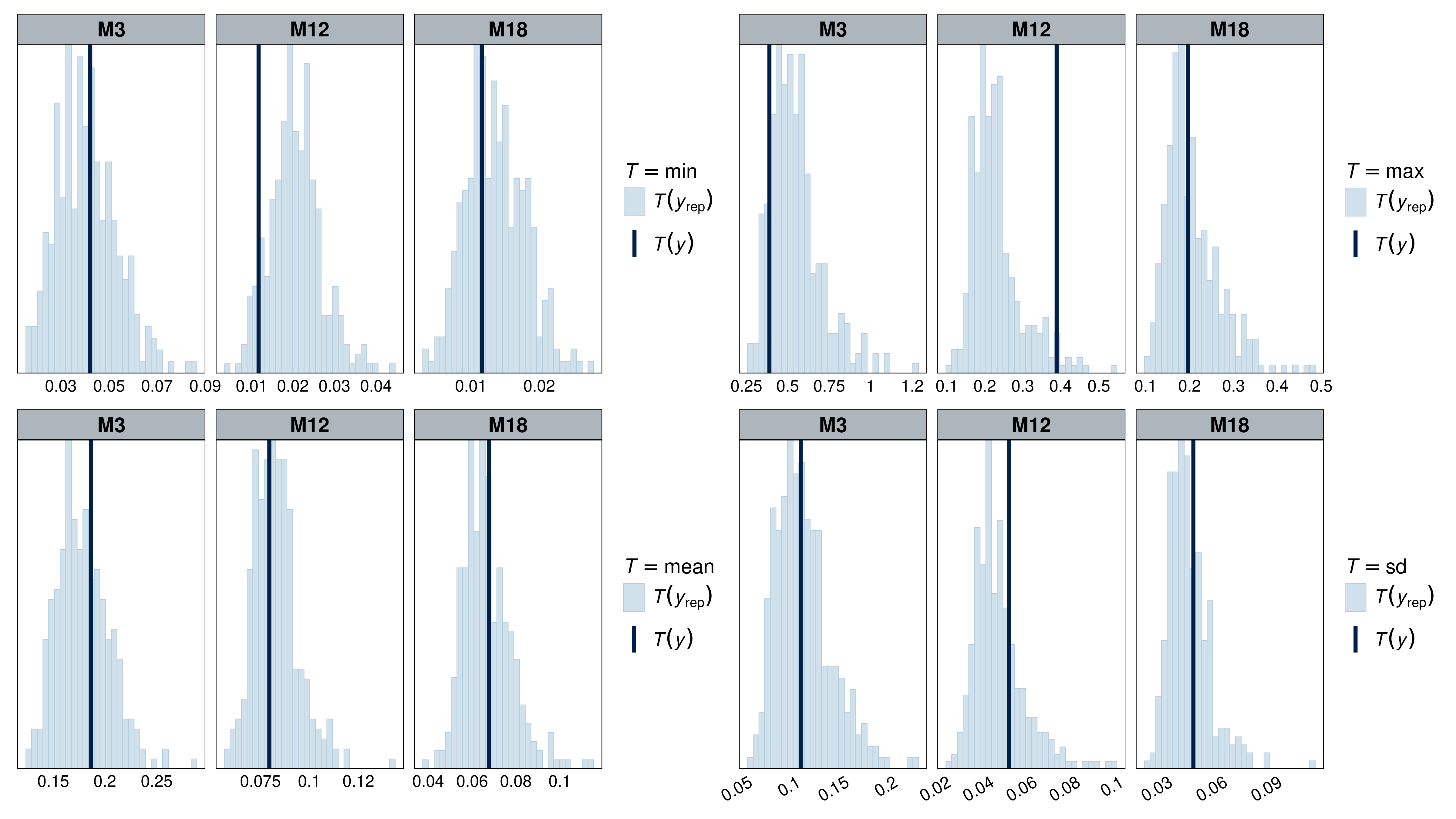
Hypotheses
Contrasts for hypotheses of interest were tested with emmeans (Lenth, 2022), with the Holm method to adjust for multiple comparisons.
Since Reelin neurons have high metabolism, we hypothesized a higher Bnip3 expression in the soma of LEC/MEC neurons in AD rats, and the difference should be higher in older rats. We used the ‘Soma’ model to test these hypotheses.
emmeans(mod_soma, specs = "genotype", type = "response") |>
contrast(method = "pairwise", adjust = "holm", infer = TRUE) contrast ratio SE df asymp.LCL asymp.UCL null z.ratio p.value
WT / McGill 0.943 0.202 Inf 0.619 1.44 1 -0.274 0.7842
Results are averaged over the levels of: age, area
Confidence level used: 0.95
Intervals are back-transformed from the log scale
Tests are performed on the log scale 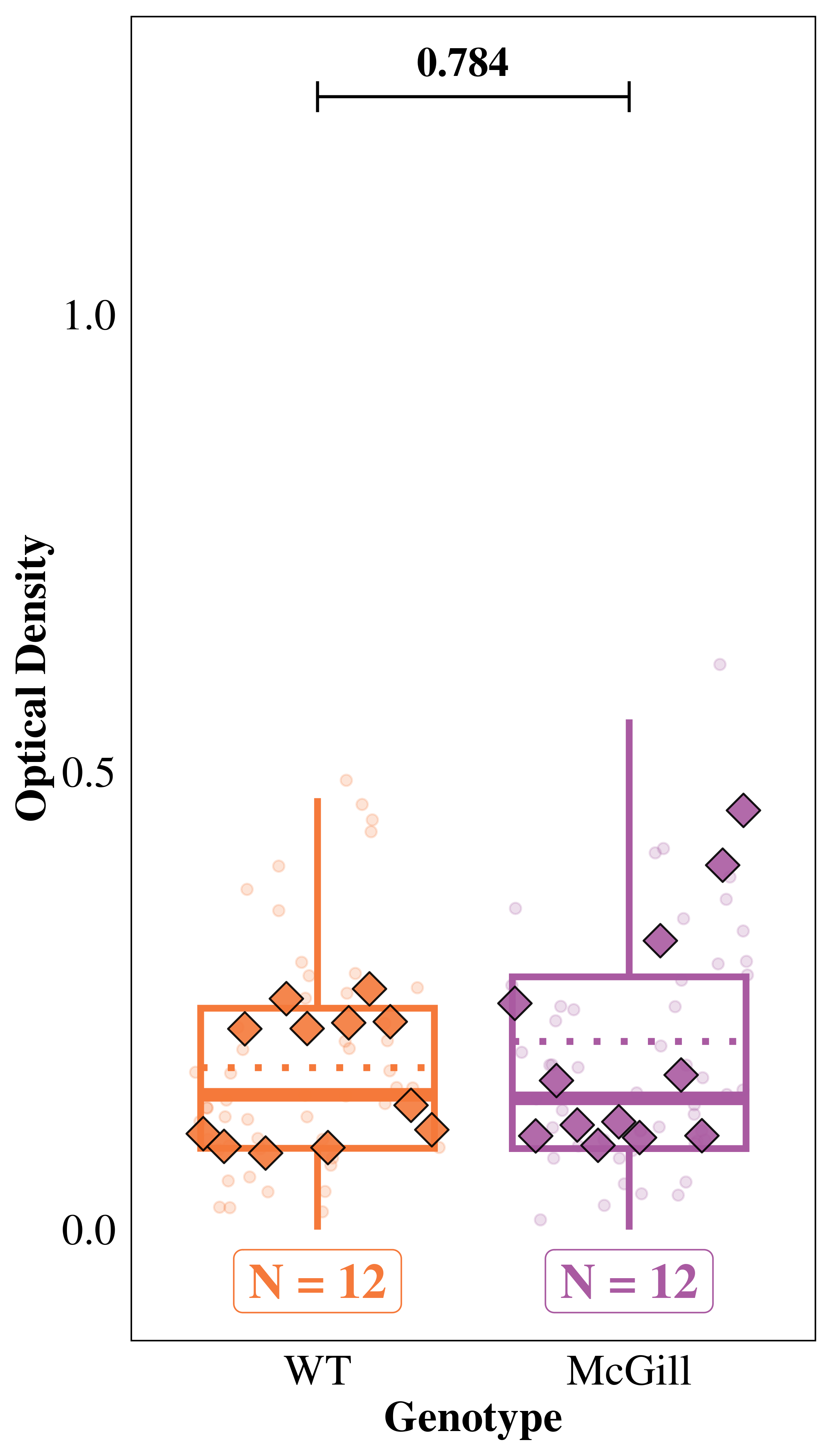
emmeans(mod_soma, specs = ~ genotype | age, type = "response") |>
contrast(method = "pairwise", adjust = "none", infer = TRUE) |>
as.data.frame() |>
modify_at("p.value", \(x) p.adjust(x, method = "holm"))age = M3:
contrast ratio SE df asymp.LCL asymp.UCL null z.ratio p.value
WT / McGill 0.5012 0.1862 Inf 0.2420 1.038 1 -1.859 0.1891
age = M12:
contrast ratio SE df asymp.LCL asymp.UCL null z.ratio p.value
WT / McGill 1.5831 0.5882 Inf 0.7643 3.279 1 1.236 0.4326
age = M18:
contrast ratio SE df asymp.LCL asymp.UCL null z.ratio p.value
WT / McGill 1.0566 0.3927 Inf 0.5100 2.189 1 0.148 0.8822
Results are averaged over the levels of: area
Confidence level used: 0.95
Intervals are back-transformed from the log scale
Tests are performed on the log scale 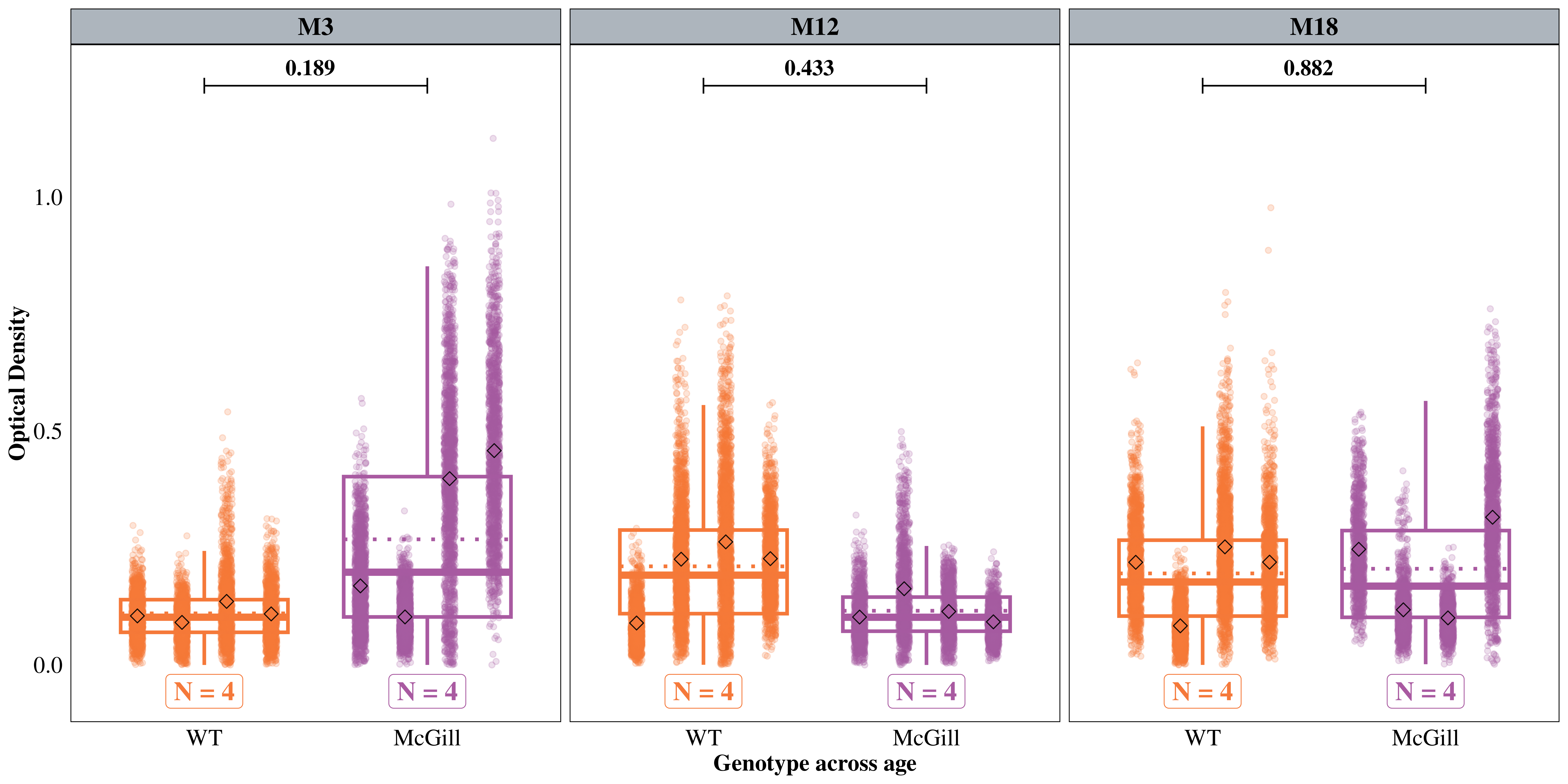
emmeans(mod_soma, specs = ~ genotype | age, type = "response") |>
contrast(interaction = "pairwise", by = NULL, adjust = "none", infer = TRUE) |>
as.data.frame() |>
modify_at("p.value", \(x) p.adjust(x, method = "holm")) genotype_pairwise age_pairwise ratio SE df asymp.LCL asymp.UCL null z.ratio p.value
WT / McGill M3 / M12 0.3166 0.1664 Inf 0.1131 0.887 1 -2.189 0.0858
WT / McGill M3 / M18 0.4744 0.2493 Inf 0.1694 1.329 1 -1.419 0.3118
WT / McGill M12 / M18 1.4983 0.7874 Inf 0.5349 4.197 1 0.769 0.4417
Results are averaged over the levels of: area
Confidence level used: 0.95
Intervals are back-transformed from the log scale
Tests are performed on the log scale 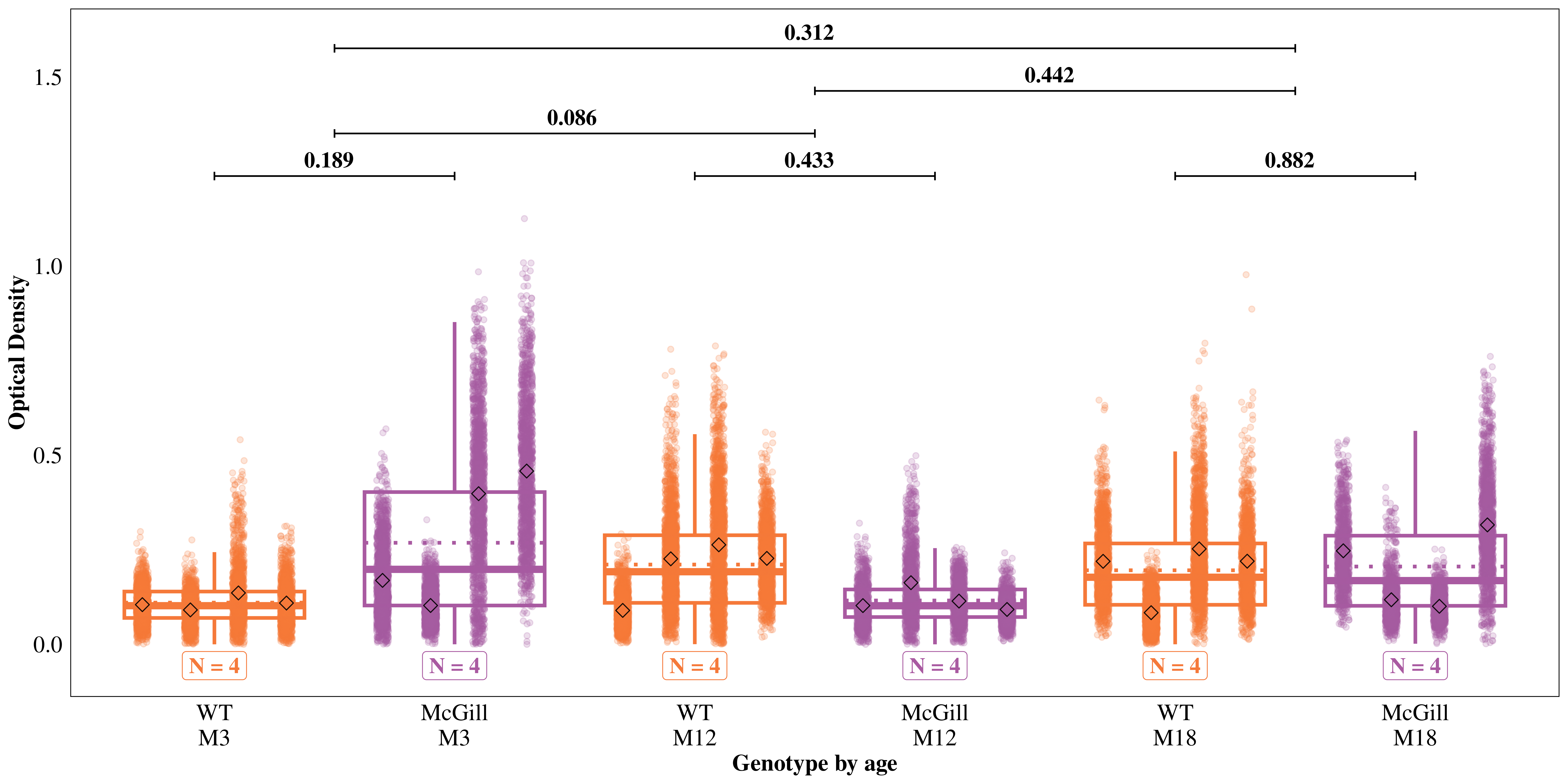
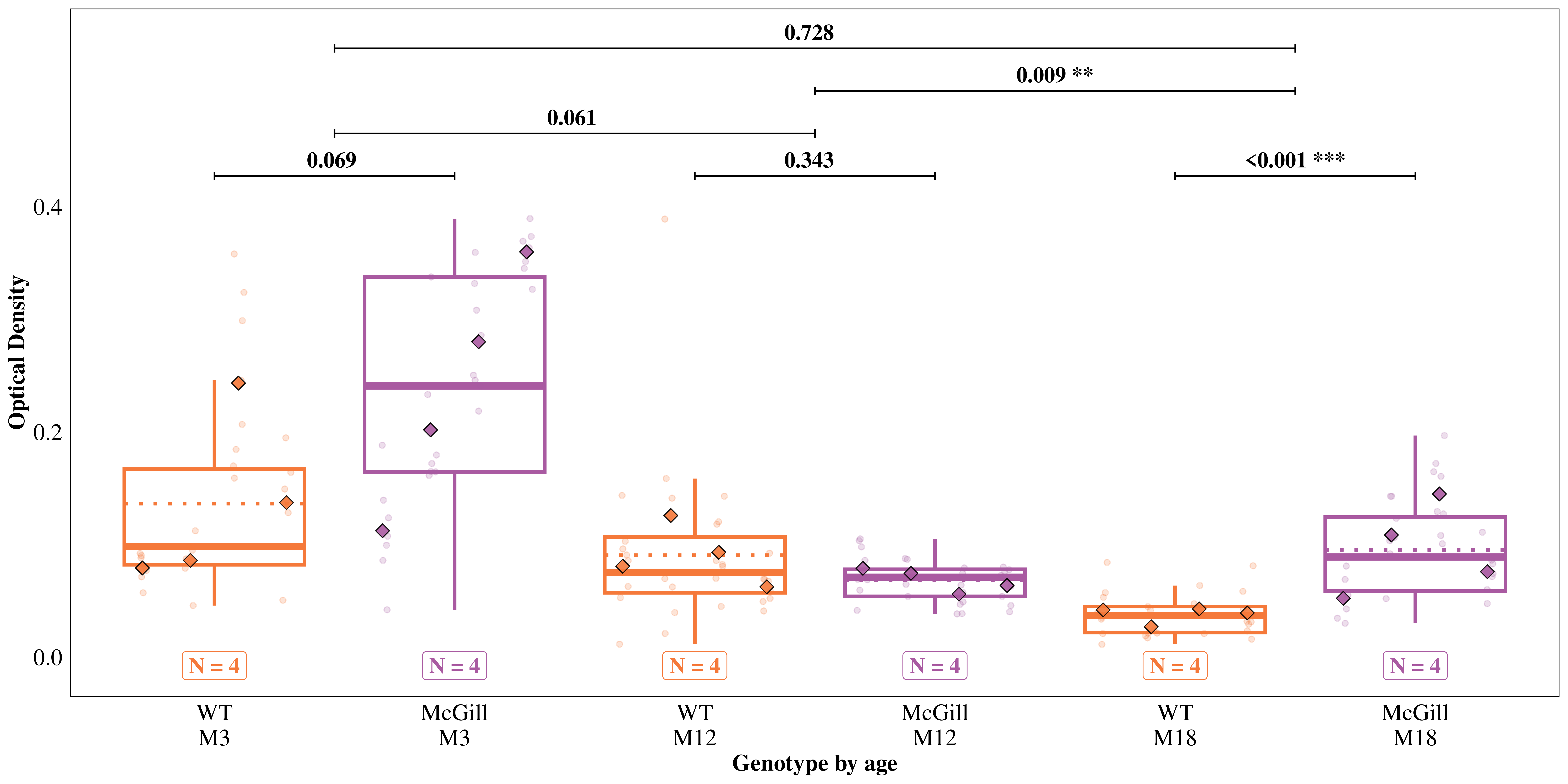
We hypothesized that BNIP3 expression would be relatively low in other areas, but may be high in the projection areas of those neurons (CA3 & dentate gyrus). We used the ‘Molecular Layer’ model to test these hypotheses.
emmeans(mod_ml, specs = "genotype", type = "response") |>
contrast(method = "pairwise", adjust = "holm", infer = TRUE) contrast ratio SE df asymp.LCL asymp.UCL null z.ratio p.value
WT / McGill 0.715 0.0999 Inf 0.543 0.94 1 -2.403 0.0163
Results are averaged over the levels of: age
Confidence level used: 0.95
Intervals are back-transformed from the log scale
Tests are performed on the log scale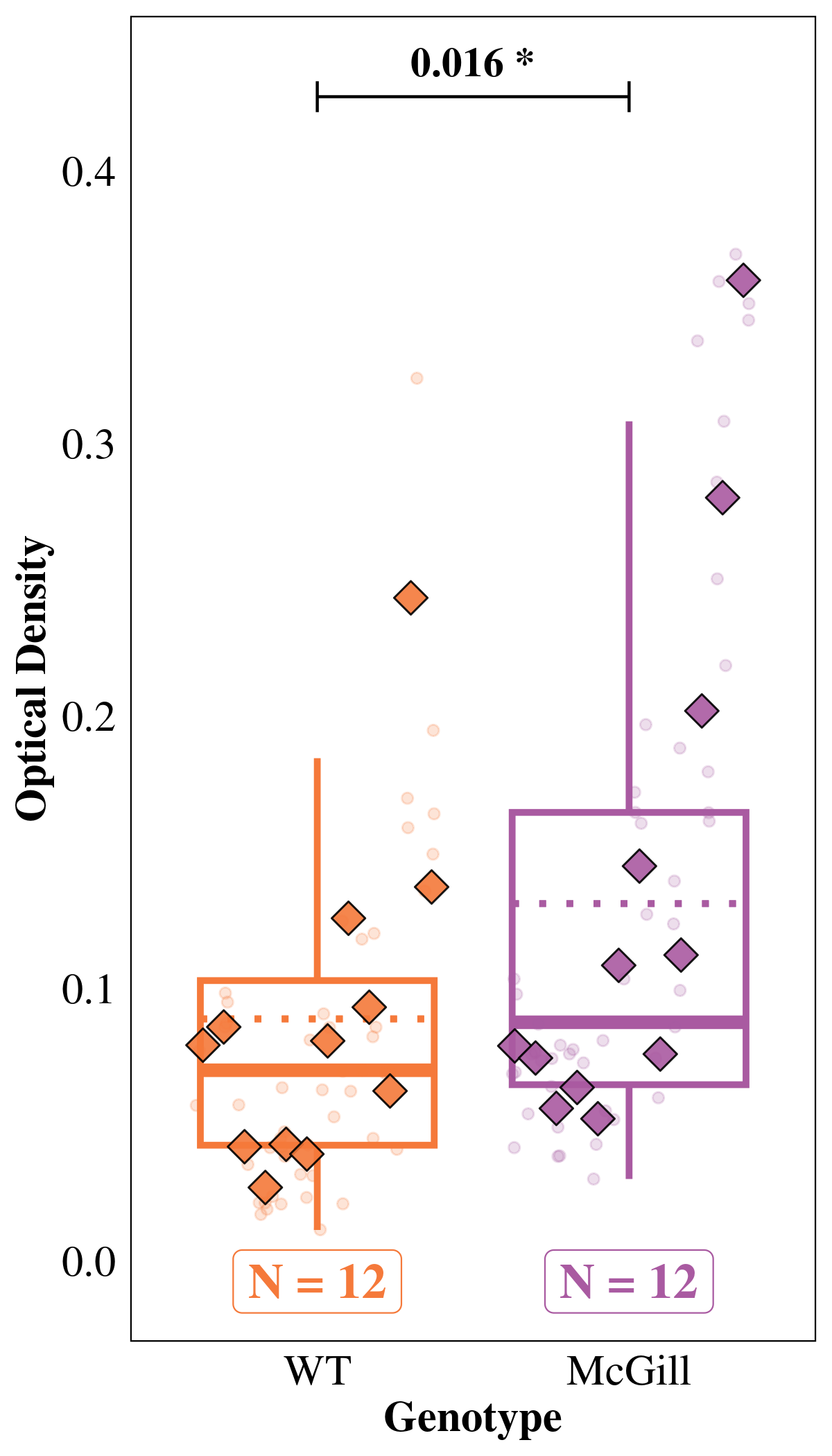
emmeans(mod_ml, specs = ~ genotype | age, type = "response") |>
contrast(method = "pairwise", adjust = "none", infer = TRUE) |>
as.data.frame() |>
modify_at("p.value", \(x) p.adjust(x, method = "holm"))age = M3:
contrast ratio SE df asymp.LCL asymp.UCL null z.ratio p.value
WT / McGill 0.5610 0.1535 Inf 0.3281 0.9592 1 -2.112 0.0694
age = M12:
contrast ratio SE df asymp.LCL asymp.UCL null z.ratio p.value
WT / McGill 1.2950 0.3529 Inf 0.7591 2.2091 1 0.949 0.3429
age = M18:
contrast ratio SE df asymp.LCL asymp.UCL null z.ratio p.value
WT / McGill 0.5021 0.0823 Inf 0.3641 0.6924 1 -4.202 0.0001
Confidence level used: 0.95
Intervals are back-transformed from the log scale
Tests are performed on the log scale 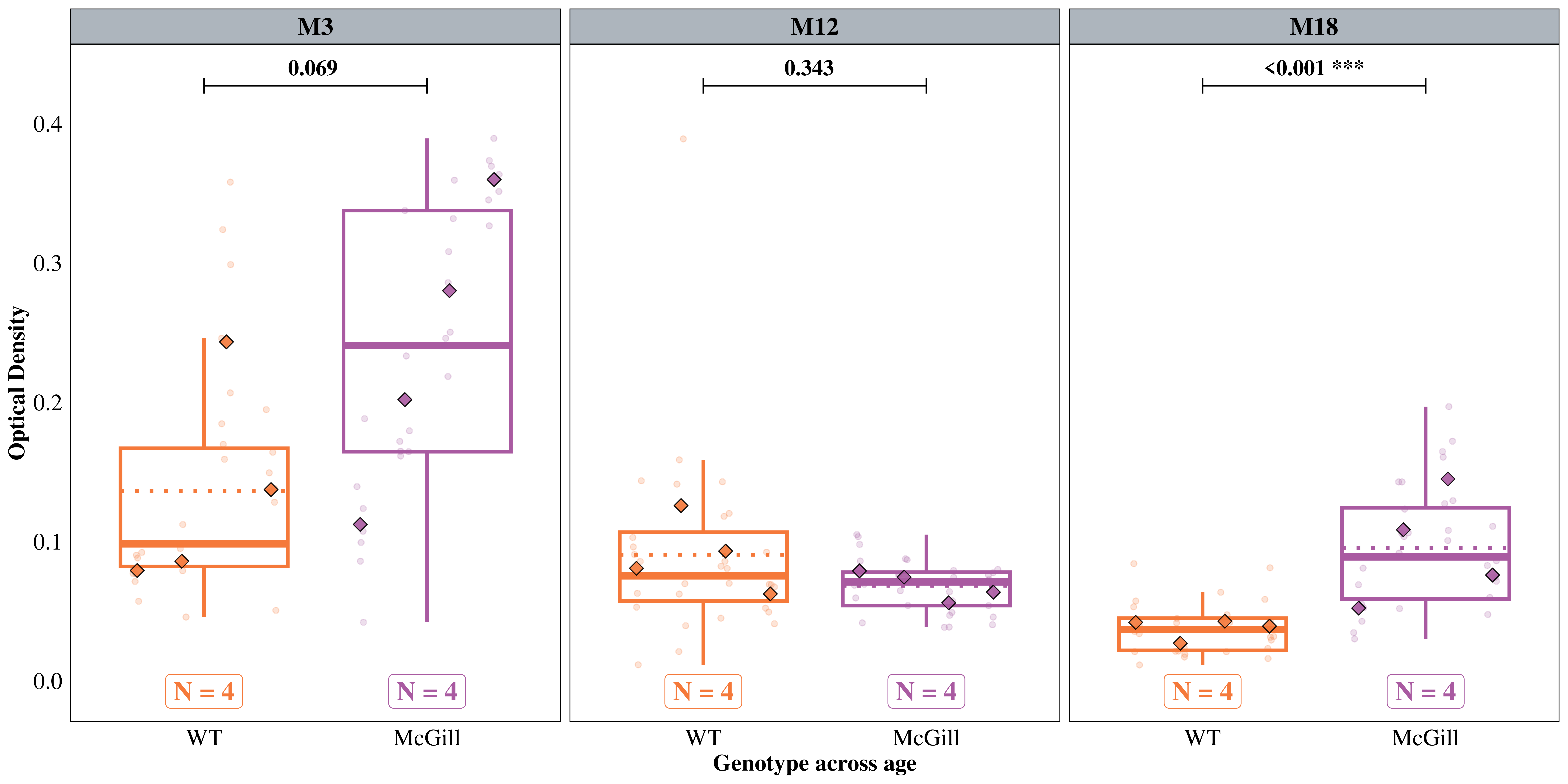
emmeans(mod_ml, specs = ~ genotype | age, type = "response") |>
contrast(interaction = "pairwise", by = NULL, adjust = "none", infer = TRUE) |>
as.data.frame() |>
modify_at("p.value", \(x) p.adjust(x, method = "holm")) genotype_pairwise age_pairwise ratio SE df asymp.LCL asymp.UCL null z.ratio p.value
WT / McGill M3 / M12 0.4332 0.1673 Inf 0.2032 0.924 1 -2.166 0.0606
WT / McGill M3 / M18 1.1172 0.3564 Inf 0.5978 2.088 1 0.347 0.7283
WT / McGill M12 / M18 2.5788 0.8202 Inf 1.3826 4.810 1 2.979 0.0087
Confidence level used: 0.95
Intervals are back-transformed from the log scale
Tests are performed on the log scale